ChemEmbed: a deep learning framework for metabolite identification using enhanced MS/MS data and multidimensional molecular embeddings
Times cited: 3
Faizan-Khan, M, Giné, R, Badia, JM, Pérez-Ribera, M, Ruiz-Botella, M, Junza, A, Capellades, J, Pérez-López, I, Xing, S, Patan, A, Brugnara, L, Novials, A, Servitja, JM, Vinaixa, M, Dorrestein, PC, Sales-Pardo, M, Guimerà, R, Yanes, O.
Brief. Bioinf.
27 (1)
,
bbag054
(2026).
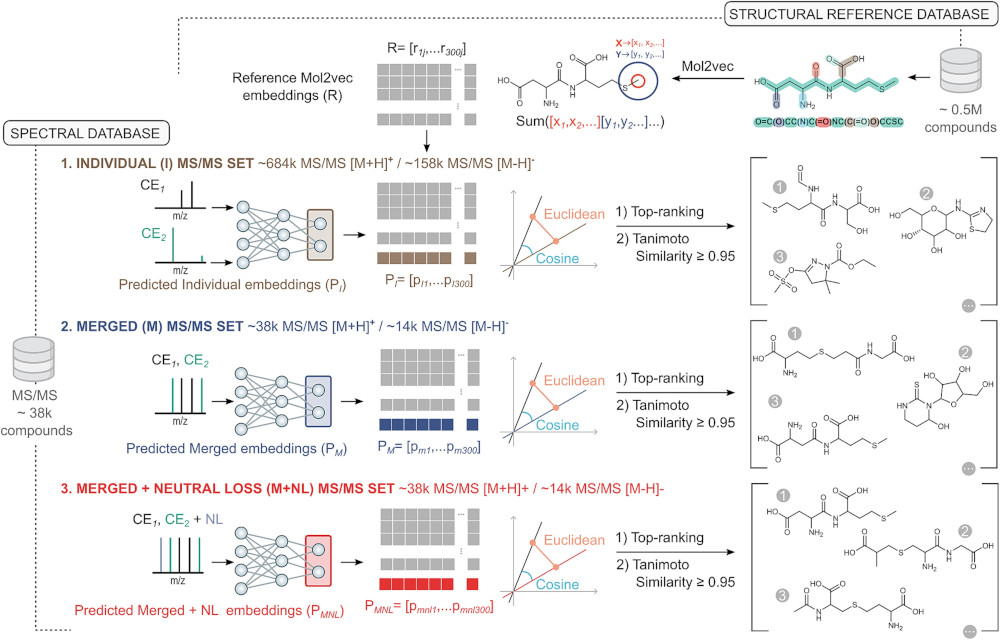
Machine learning offers a promising path to annotating the large number of unidentified MS/MS spectra in metabolomics, addressing the limited coverage of current reference spectral libraries. However, existing methods often struggle with the high dimensionality and sparsity of MS/MS spectra and metabolite structures. ChemEmbed tackles these challenges by integrating multidimensional, continuous vector representations of chemical structures with enhanced MS/MS spectra. This enhancement is achieved by merging spectra across multiple collision energies and incorporating calculated neutral losses from 38 472 distinct compounds, providing richer input for a convolutional neural network (CNN). ChemEmbed ranks the correct candidate first in over 42% of cases and within the top five in more than 76% of cases. In external benchmarks such as CASMI 2016 and 2022, ChemEmbed outperforms SIRIUS 6, the current state-of-the-art in computational metabolomics. We applied ChemEmbed to predict structures in the Annotated Recurrent Unidentified Spectra (ARUS) dataset and confirmed 25 previously unidentified compounds. These findings demonstrate ChemEmbed’s potential as a robust, scalable tool for accelerating metabolite identification in untargeted mass spectrometry workflows.